In February 2025, an infant named KJ — born with a rare metabolic disorder called CPS1 deficiency that would have been fatal without constant medical management — received a customised CRISPR gene-editing therapy developed specifically for him at Children’s Hospital of Philadelphia. The therapy was designed, manufactured, and delivered in approximately six months after his diagnosis. KJ is now growing well and thriving. He is the first person in history to be treated with a bespoke, individualised CRISPR therapy — a therapy that could not have existed for any patient before him, designed for the specific genetic mutation in his specific cells, administered in a single infusion. As the researchers who built it put it: the challenge now is “to go from CRISPR for one to CRISPR for all.”
That challenge captures exactly where gene editing stands in 2026. The science has progressed from laboratory curiosity through laboratory tool through approved medicine to personalised therapeutics in roughly a decade — a pace of clinical translation without precedent in the history of molecular biology. Casgevy, the first CRISPR-based medicine ever approved, received FDA authorisation in December 2023 and has since been approved in the US, UK, EU, Canada, Switzerland, and multiple Middle Eastern countries. Its clinical results are historic: 100 percent of sickle cell disease patients in its pivotal trial achieved freedom from the agonising vaso-occlusive crises that define the disease, with a mean crisis-free period extending beyond 35 months. For transfusion-dependent beta-thalassaemia, 98.2 percent of patients achieved transfusion independence. For diseases that have caused suffering for centuries, these are not incremental improvements. They are functional cures.
Approximately 250 clinical trials involving CRISPR-based gene-editing therapies are currently monitored globally, with more than 150 actively enrolling or treating patients. The conditions under investigation span blood disorders, cardiovascular disease, cancer, hereditary blindness, metabolic disorders, and rare genetic diseases. Base editing — a more precise variant of CRISPR that rewrites individual DNA letters without cutting the double helix — is entering clinical trials for conditions including chronic granulomatous disease and liver diseases, and its developers won the 2025 Breakthrough Prize in Life Sciences. Prime editing, still more precise, is moving toward early clinical testing. The FDA released draft guidance in February 2026 creating a “plausible mechanism framework” for platform therapies — a regulatory pathway that could enable a single clinical trial to test a platform customisable for individual patients, directly addressing the scalability challenge that KJ’s case illustrates.
This guide explains how CRISPR works, what Casgevy’s results mean, the new generation of more precise editing tools, the most significant clinical programmes underway, the delivery challenge that determines whether gene editing can scale from wealthy patients at specialist centres to anyone who needs it, and the ethical questions that the technology’s expanding capabilities are forcing society to address.
How CRISPR Works: The Molecular Scissors Explained
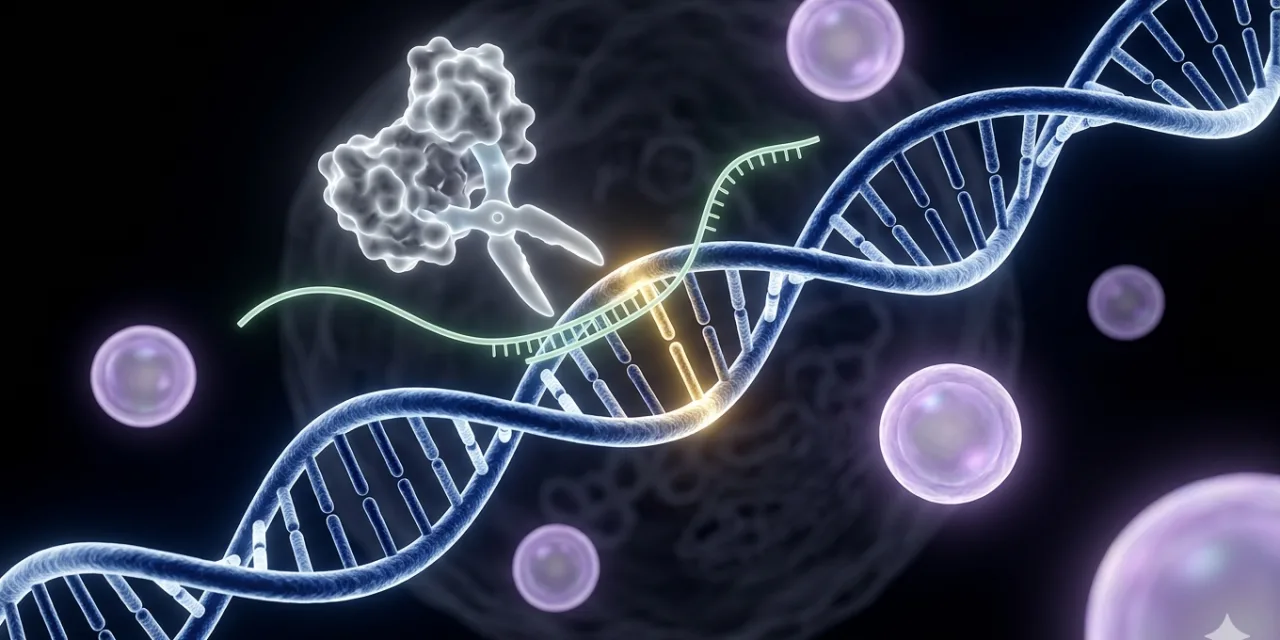
CRISPR — Clustered Regularly Interspaced Short Palindromic Repeats — was discovered not in a biotech lab but in bacteria. Bacteria use CRISPR as an immune system: when they encounter viral DNA, they store fragments of it in their genome as CRISPR sequences, and when the same virus attacks again, they deploy CRISPR-associated proteins (Cas proteins) that recognise the stored viral sequence and cut the viral DNA apart. Researchers Jennifer Doudna and Emmanuelle Charpentier — who shared the 2020 Nobel Prize in Chemistry for the work — realised that this bacterial immune system could be reprogrammed: by providing a synthetic guide RNA matching any sequence of interest, the Cas9 protein could be directed to cut DNA at essentially any location in any genome.
The operational mechanism is elegantly simple. A guide RNA — a short molecule designed to match the DNA sequence at the target location — brings the Cas9 protein to the precise spot in the genome where an edit is needed. The Cas9 protein makes a precise cut through both strands of the DNA double helix at that location. The cell’s own DNA repair machinery then takes over: it can either join the cut ends imprecisely (which typically disrupts or disables the gene — useful for eliminating disease-causing gene activity) or use a provided DNA template to repair the cut in a precise, specified way (which can correct a disease-causing mutation to the healthy sequence). The guide RNA sequence is the variable component; it can be designed in a matter of days on a computer to target virtually any location in any genome. Everything else — the Cas9 protein, the delivery vehicle, the dosing framework — remains essentially constant across different therapeutic applications.
This is the “platform” character of CRISPR that makes it so scientifically and commercially significant: the same fundamental machinery can be reprogrammed for a different target with only the guide RNA sequence changed. Unlike conventional drug development, where each new disease target requires developing an entirely new molecular entity from scratch, CRISPR-based therapies for different genetic diseases are variations on the same platform. This is what makes the FDA’s February 2026 platform therapy framework potentially transformative: if regulators accept that a platform with a demonstrated safety record can be customised for individual patients without each customisation requiring a full new drug approval, the path from a patient’s diagnosis to a bespoke therapy compresses dramatically.
Casgevy: What Functional Cures Look Like in Practice
Casgevy works by reactivating fetal haemoglobin — a form of haemoglobin that fetuses produce in the womb but that is normally switched off after birth in favour of adult haemoglobin. In sickle cell disease, the adult haemoglobin gene carries a mutation that causes haemoglobin to crystallise under low-oxygen conditions, distorting red blood cells into a sickle shape that blocks blood vessels and causes the excruciating vaso-occlusive crises that characterise the disease. In transfusion-dependent beta-thalassaemia, the adult haemoglobin gene is either absent or non-functional, requiring patients to receive regular blood transfusions to survive. In both cases, fetal haemoglobin can do the job that mutant adult haemoglobin cannot — if it can be reactivated in adult cells.
Casgevy uses CRISPR to edit patients’ own haematopoietic stem cells — the blood-forming cells in the bone marrow — by inactivating a gene called BCL11A that normally suppresses fetal haemoglobin production in adults. The edited stem cells are then reinfused into the patient after chemotherapy depletes the existing bone marrow population, allowing the edited cells to repopulate the marrow and produce fetal haemoglobin. The discovery that BCL11A is the key suppressor of fetal haemoglobin — the finding that earned Stuart Orkin a share of the 2026 Breakthrough Prize in Life Sciences — is what made Casgevy possible. The underlying science pointed directly from basic genetic research to a specific therapeutic target.
The clinical results in the pivotal CLIMB-121 and long-term follow-up CLIMB-131 trials are the most compelling gene therapy outcomes ever documented at scale. All 45 sickle cell disease patients enrolled achieved complete freedom from vaso-occlusive crises at 12 months. For beta-thalassaemia, 55 of 56 patients (98.2 percent) achieved transfusion independence. These patients had previously faced a lifetime of recurrent crises requiring hospitalisation, or lifetime dependence on blood transfusions — and now do not. The durability of the results, with follow-up extending beyond three years, suggests that the edits are permanent and stable in the repopulated marrow.
The gap between these scientific results and real-world access, however, is substantial. As of early 2026, only approximately 60 patients have been treated with Casgevy worldwide. The treatment process requires apheresis to collect stem cells, CRISPR editing at a specialised manufacturing facility, several weeks of chemotherapy conditioning to deplete the patient’s existing bone marrow, reinfusion of edited cells, and months of recovery in a medical centre with haematopoietic stem cell transplant capability. The list price in the United States is approximately $2.2 million per treatment — making it among the most expensive medical interventions in history. Sickle cell disease disproportionately affects people of African descent, and beta-thalassaemia disproportionately affects people of Mediterranean, Middle Eastern, and South and Southeast Asian descent — populations that are, on average, significantly less likely to have the medical infrastructure and financial resources that current Casgevy delivery requires. The access problem is not peripheral to the story of CRISPR medicine — it is central to whether the technology fulfils its promise.
The Next Generation: Base Editing and Prime Editing
CRISPR-Cas9’s molecular scissors mechanism — cutting both strands of the DNA double helix — is powerful but imprecise in a specific way: the cut itself creates a biological emergency that cells respond to with DNA repair mechanisms that are not always precise. Non-homologous end joining, the cell’s most efficient repair pathway, frequently introduces small insertions or deletions at the cut site that can create off-target effects at genomic locations that incidentally share sequence similarity with the guide RNA’s target. The two next-generation editing platforms — base editing and prime editing — address this limitation by making precise nucleotide-level changes without cutting both DNA strands.
Base editing, developed in David Liu’s laboratory at the Broad Institute of MIT and Harvard in 2016, uses a modified Cas9 protein that has been disabled from cutting DNA and instead serves as a precise DNA-sequence navigator — bringing a chemical enzyme to the target location that directly converts one DNA letter to another in place. Cytosine base editors convert cytosine (C) to thymine (T); adenine base editors convert adenine (A) to guanine (G). Together, they can address approximately 60 percent of all known single-nucleotide disease-causing variants — the single-letter spelling mistakes in the genome that cause thousands of genetic diseases. Liu received the 2025 Breakthrough Prize in Life Sciences for this work, which transformed CRISPR from a tool for disrupting gene function into a tool for correcting genetic diseases at single-nucleotide precision. Clinical trials using base editing are already underway for haematological disorders and inherited liver diseases, and one remarkable trial demonstrated 90 percent correction of a disease-causing protein within 14 days of a single infusion — the first demonstration that CRISPR can directly correct a disease-causing mutation rather than compensating for it by disrupting a related gene.
Prime editing, also from Liu’s laboratory (2019), extends the capability further: using a reverse transcriptase enzyme fused to a modified Cas protein and a specially designed guide RNA, prime editing can make any of the 12 possible single-letter DNA changes, plus small insertions and deletions, all without cutting both DNA strands. Prime editing can theoretically correct approximately 90 percent of known disease-causing variants. Its clinical translation is more recent than base editing — the first prime editing trials are expected to begin enrolling patients in 2026 — but its precision advantage over first-generation CRISPR makes it the leading candidate for disease applications where off-target effects would be clinically unacceptable.
Cardiovascular Disease: CRISPR’s Next Major Frontier
Blood disorders were the natural first targets for CRISPR medicine because the relevant cells — haematopoietic stem cells — can be removed from the patient, edited outside the body, and reinfused, avoiding the delivery challenges that complicate in-body editing. Cardiovascular disease is the next major frontier, and the approach being pursued is conceptually audacious: permanently editing the PCSK9 gene in liver cells to eliminate its activity and reduce LDL cholesterol for life with a single treatment.
PCSK9 is a protein that degrades the receptors that clear LDL cholesterol from the blood. People with natural loss-of-function variants of PCSK9 have significantly lower LDL levels and dramatically reduced risk of heart attack and stroke throughout their lives, with no apparent adverse health effects — demonstrating that the gene is a safe and effective cardiovascular risk target. Verve Therapeutics initiated a phase I clinical trial using base editing to inactivate PCSK9 in liver cells — delivered via lipid nanoparticles (LNPs) administered intravenously, the same delivery platform used in mRNA COVID-19 vaccines. Early results showed LDL reductions of approximately 50 percent that appear durable — consistent with the expectation that a permanent edit to a non-dividing liver cell would maintain its effect for life. Chinese biotech AccurEdit Therapeutics reported similar 50 percent LDL reductions in their PCSK9 editing trial in 2024, with no severe adverse reactions.
The cardiovascular CRISPR programme, if it reaches approval, would represent a fundamental shift in how heart disease is treated: from lifetime daily medication (with its adherence challenges, side effects, and cumulative costs) to a single infusion that permanently reduces a key risk factor. The addressable market is enormous — elevated LDL is among the most prevalent cardiovascular risk factors globally — and the commercial potential is driving significant investment in perfecting the in vivo delivery technology.
The Delivery Problem: The Difference Between Lab Results and Patient Access
The most important technical challenge limiting CRISPR’s clinical impact in 2026 is not the editing itself — guide RNA design, Cas protein engineering, and editing precision have all advanced dramatically — but delivery: how to get the editing components into the right cells in the right tissue efficiently, safely, and with minimal off-target activity in tissues where editing is not intended.
The current approaches divide into ex vivo (editing outside the body) and in vivo (editing inside the body) strategies. Ex vivo editing — used for Casgevy — removes target cells from the patient, edits them in a laboratory setting, and reinfuses them. This provides maximum control over editing conditions and allows extensive quality testing before administration, but it is logistically complex, expensive, requires specialist medical infrastructure, and is only feasible for cell types (primarily blood cells and immune cells) that can be practically harvested, edited, and reinfused.
In vivo editing — delivering CRISPR components directly into the body to edit cells in their native tissue — is where the most significant ongoing research investment is concentrated, because it would remove most of the barriers to scale. If editing components can be packaged and delivered to specific tissues without ex vivo cell handling, the treatment process could eventually resemble a standard intravenous infusion rather than a months-long medical procedure.
Lipid nanoparticles (LNPs) are the current leading in vivo delivery platform for CRISPR components, particularly for liver-targeting applications. LNPs naturally accumulate in the liver after intravenous administration — the same property exploited in COVID-19 mRNA vaccines — making them highly effective for editing liver cells while having limited off-target distribution to other organs. The cardiovascular PCSK9 programmes use LNP delivery for exactly this reason. Extending in vivo delivery to other tissues — muscle, lung, brain, eye, kidney — requires engineering LNPs or alternative delivery vehicles (such as adeno-associated virus vectors) that preferentially target each tissue. Significant progress has been made for some targets; others remain technically challenging.
Cancer: CRISPR Meets Oncology
The application of CRISPR to cancer is proceeding along multiple distinct approaches, each exploiting different aspects of the technology’s capabilities. The most clinically advanced approach involves engineering T cells — the immune system’s cancer-fighting cells — to be more effective at identifying and killing tumour cells. CRISPR is used to remove genes that limit T cell activity (including genes that cause T cell exhaustion and genes that make T cells susceptible to tumour suppression signals) and in some cases to add genes that target T cells toward specific tumour markers. These CRISPR-engineered T cells are then expanded and reinfused in a process conceptually related to the chimeric antigen receptor T cell (CAR-T) therapies that have transformed haematological cancer treatment.
A distinct cancer application uses CRISPR to create “off-the-shelf” or allogeneic cell therapies — engineered immune cells derived from healthy donors rather than the patient, with CRISPR editing used to remove the genes that would cause the donor cells to attack the patient’s body (graft-versus-host disease) while preserving their cancer-killing activity. Allogeneic cell therapies, if successful, would be significantly more accessible than personalised autologous approaches because a single manufacturing batch could treat many patients — reducing cost and eliminating the weeks-long delay of creating patient-specific therapies. Multiple Phase I trials of allogeneic CRISPR-edited cell therapies are currently underway for various haematological cancers. As of 2026, more than 150 active gene editing clinical trials include cancer as a target indication.
The Ethics: From Treatment to Enhancement
The ethical landscape of CRISPR expanded dramatically in 2018, when Chinese scientist He Jiankui announced that he had used CRISPR to edit human embryos that were subsequently implanted and brought to term — producing the first gene-edited human beings, twin girls with edits intended to confer resistance to HIV infection. He Jiankui was widely condemned by the scientific community, imprisoned by Chinese authorities for three years, and remains a cautionary example of what happens when scientific capability races ahead of ethical consensus and regulatory oversight. His experiment did not produce a verifiable medical benefit, introduced untested heritable changes into the human germline, and was conducted without appropriate informed consent or institutional oversight.
The He Jiankui case crystallised the central ethical distinction that governs CRISPR in medicine: somatic editing versus germline editing. Somatic editing changes the DNA of specific cells in a living person and affects only that person — exactly what Casgevy does. The edits are not inherited by the patient’s children. Germline editing changes the DNA of an embryo, egg, sperm, or early embryo, and the changes are transmitted to all cells in the resulting person and to their descendants indefinitely. The scientific community, regulatory agencies, and ethical consensus broadly support somatic editing for therapeutic purposes (with appropriate clinical oversight). Germline editing for therapeutic or enhancement purposes in humans remains broadly opposed as premature — the long-term heritable effects of genomic changes cannot be predicted with current knowledge, and the changes cannot be reversed once inherited.
The enhancement question is where the ethical debate extends beyond the immediate clinical present. CRISPR’s precision means that, as the genome’s functional landscape becomes better understood, it becomes technically conceivable to edit not for disease correction but for enhancement — higher intelligence, athletic capability, resistance to common diseases, extended lifespan. These applications are speculative rather than imminent: the genetic architecture of complex traits like intelligence or athletic performance is enormously complex, involving thousands of variants with small, context-dependent effects that current science does not understand well enough to edit productively. But the trajectory of the technology and the knowledge base makes the question not merely hypothetical, and the societal frameworks for governing it — who decides what edits are permissible, who has access to enhancements that are permitted, and how heritable enhancements affect human equality — are not yet developed to the level of sophistication the technology will eventually require.
What 2026 Means for the Future of CRISPR Medicine
The pattern of CRISPR’s clinical development in 2026 suggests a technology transitioning from proof-of-concept to systematic clinical deployment, at a pace limited less by science than by the practical challenges of manufacturing, delivery, cost, and regulatory frameworks. Several near-term milestones are visible. The FDA’s February 2026 platform therapy guidance framework, if implemented, will significantly accelerate the regulatory path for personalised CRISPR therapies — potentially enabling bespoke treatments like KJ’s to be developed and delivered within months rather than years for patients with rare genetic diseases that no existing treatment addresses.
The number of approved CRISPR therapies is expected to grow from one (Casgevy) to potentially a dozen or more by 2030, with blood disorders remaining the near-term dominance but liver diseases, cardiovascular conditions, hereditary blindness, and certain cancers all in advanced clinical development. The transition from ex vivo to in vivo delivery will be the most consequential technical development — successful in vivo delivery to non-liver tissues would open the full range of genetic diseases to CRISPR treatment while dramatically reducing procedural complexity and cost.
The access challenge remains the most important non-scientific constraint on the technology’s impact. Current CRISPR therapies are available almost exclusively at specialist centres in high-income countries, at price points that make them accessible only through exceptional insurance coverage or clinical trial enrolment. The vision of CRISPR as a medicine for patients worldwide — not merely wealthy patients at specialist centres — requires advances in manufacturing scale, cost reduction, simplified delivery protocols, and international regulatory coordination that the scientific advances alone cannot provide. Decentralised manufacturing models — allowing cell therapies to be produced closer to the patients who need them — are being explored specifically to address this gap, as demonstrated by the Boston Children’s Hospital collaboration with Caring Cross announced in early 2026.
The code of life is being rewritten. For the first time in the history of medicine, clinicians have a general-purpose tool that can, in principle, correct virtually any genetic disease at its molecular root rather than managing symptoms throughout a patient’s lifetime. The first functional cures are already in patients. The next decade of CRISPR medicine will determine how broadly, how safely, and how equitably that capability reaches the people who need it most.






0 Comments
No comments yet. Be the first to share your thoughts!